Sjukdomen med hundra ansikten
Text: Christian Hellman
Retinitis pigmentosa (RP) är en komplex sjukdom att beskriva. Beroende på vilken definition man väljer, kan RP betraktas som en grupp av ett hundratal likartade sjukdomar eller som en sjukdom som förekommer i ett hundratal olika former. Gemensamt för alla former är att de är ärftliga, uppstår genom mutationer i ett stort antal gener och drabbar syncellerna i näthinnan.
Flera av de stora ögonsjukdomarna, såsom glaukom och åldersrelaterad makuladegeneration, drabbar främst äldre personer. RP kan drabba både unga och gamla och är därför en av de vanligaste orsakerna till blindhet bland personer i arbetsför ålder. Sjukdomen är ändå förhållandevis sällsynt, med en förekomst på ungefär 1 av 4 000 personer globalt.
Många former med olika sjukdomsförlopp
Det finns inga tydliga yttre faktorer som påverkar risken för att utveckla retinitis pigmentosa. Sjukdomen är helt och hållet genetiskt betingad, vilket innebär att en familjehistorik av RP utgör den största riskfaktorn. Om en familjemedlem har sjukdomen, är risken större att man själv får den. Enligt ögonläkaren Henrik Bygglin är inte allt ändå förutbestämt.
– Sjukdomen kan också drabba olika medlemmar av samma familj på olika sätt och med varierande svårighetsgrad. I sällsynta fall kan den uppstå även om det inte finns någon familjehistorik alls, förklarar han.
Symtomen är i regel mycket allvarliga, men det är ändå ovanligt att sjukdomen leder till total blindhet. De många olika formerna ger upphov till stora individuella variationer i både symtom och sjukdomsförlopp. Somliga upplever en långsam försämring av synen under flera decennier, medan andra förlorar synförmågan mycket snabbare.
– Sjukdomen dyker ofta upp redan i barndomen eller tonåren, men de första symtomen kan vara så pass lindriga att de går oupptäckta i flera år, berättar Bygglin.
Angriper stavar och tappar
Klassisk och omvänd retinitis pigmentosa är de två vanligaste formerna av sjukdomen. En grundläggande skillnad mellan dessa är att de angriper syncellerna i olika ordningsföljd.
Näthinnan innehåller två typer av synceller: stavar och tappar. Stavarna är mycket ljuskänsliga, men kan inte urskilja färger. De ligger i utkanten av näthinnan och ansvarar för mörkerseendet och det perifera synfältet. Tapparna registrerar färger, men är inte särskilt ljuskänsliga. De ligger tätt samlade i mitten av näthinnan och ansvarar för färgseendet och synskärpan i det centrala synfältet.
Vid klassisk retinitis pigmentosa drabbas stavarna först. Det första symtomet är därför ofta nedsatt mörkerseende. Det blir också allt svårare för ögonen att ställa om från ljus till mörker, vilket leder till att man lätt blir bländad. Med tiden krymper synfältet från kanterna, tills man till slut ser omvärlden som genom ett smalt rör. När sidoseendet försvunnit helt, kallas detta för tunnel- eller kikarseende.
– När små fläckar börjar uppstå i synfältet, är det ett tecken på att även tapparna drabbats. Med tiden växer fläckarna sig större, och i avancerade stadier kan även det centrala seendet försämras kraftigt, förklarar Bygglin.
Omvänd RP utgör cirka 20-30 % av alla fall. Som namnet antyder är sjukdomsförloppet omvänt, jämfört med den klassiska formen. Tapparna drabbas först, medan stavarna kan fortsätta att fungera i många år. Vid omvänd RP börjar därför symtomen med bortfall av det centrala seendet.
Diagnosprocessen
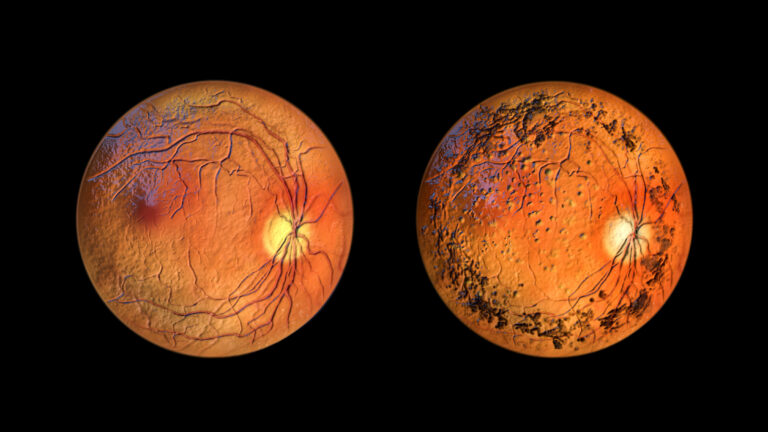
Retinitis pigmentosa utvecklas långsamt och symtomen kan vara otydliga i början av sjukdomsförloppet. Därför tar det ofta tid att ställa en korrekt diagnos. Ögonläkaren börjar vanligen med att undersöka hur mycket av synfältet som är intakt. Näthinnans respons på ljus mäts med elektroretinografi (ERG). Fundusfotografering är ett annat viktigt diagnostiskt verktyg, där man tar bilder av näthinnan för att identifiera typiska förändringar, såsom de pigmentansamlingar som gett sjukdomen dess namn.
– Med genetisk testning bekräftar vi diagnosen och identifierar den specifika mutation som orsakar sjukdomen. Detta är viktigt för att kunna erbjuda familjen information om sjukdomen och sannolikheten att den förs vidare, berättar Bygglin.
Hjälp och hopp finns
För närvarande finns ingen botande behandling för retinitis pigmentosa, men det pågår en intensiv forskning för att utveckla nya terapier. Det finns därför ett visst hopp om nya behandlingsalternativ inom de närmaste åren. Genterapi har potential att reparera eller ersätta de defekta generna. Med stamcellsterapi kan det bli möjligt att skapa nya synceller i näthinnan. Det finns också ett litet antal patienter som återfått en del av sin synförmåga genom retinala implantat, men tekniken är fortfarande på ett experimentellt stadium.
I dagsläget fokuserar behandlingen på att hantera symtomen och förbättra livskvaliteten. Vetenskapliga studier har visat att A-vitaminterapi i vissa fall kan bromsa sjukdomsförloppet. Hjälpmedel såsom ledarhund, specialanpassade glasögon och olika typer av appar och digitala hjälpmedel kan underlätta vardagen i betydande grad. Genom rehabiliteringsverksamhet kan man få hjälp att anpassa sig till sin synnedsättning och leva ett självständigt liv.
– Det är viktigt att diagnosen ställs så tidigt som möjligt. Då kan sjukdomsförloppet kanske bromsas tillräckligt för att patienten ska få chansen att dra nytta av framtida behandlingar, förklarar Bygglin.
Den här artikeln publicerades i Synvinkel 4/2024.
Hur är det att leva med RP i vardagen? Linn berättar: ”Alla med vit käpp är inte blinda”